H2 Deck By Bold Name
h2 xxxxxx
H1 xxxxxx
h2 xxxxx

Management
The most effective medical device organizations don’t slow innovation to achieve compliance. By Chiratana Pot
When Milestones Lie: Why Medical Device Teams Fall Out of Alignment and How to Prevent It
medical
H2 Deck Info By Paragraph Style Bold
Headline
From a regulatory standpoint, progress in medical device development is measured by evidence, not by phase completion. Yet many product development milestones still signal readiness before the supporting documentation is complete, approved, and traceable. Fragmented systems allow design documents, quality documents, manufacturing records, risk controls, and approvals to drift out of alignment with the work being done, often without immediate visibility. In a regulatory environment that increasingly favors connected, predictive quality systems, this misalignment has become one of the most common and avoidable sources of regulatory risk.
In medical device programs, it’s common for development to appear on track. Project plans show milestones advancing. Engineering teams move to the next phase. Leadership sees green status indicators.
Yet beneath that surface, quality and regulatory leaders frequently sense unease. Documentation doesn’t quite reflect the current design. Approvals trail behind decisions. Risk files lag changes already implemented.
The issue isn’t that teams “miss timelines.” The issue is that milestones advance without the evidence that regulators expect to support them. Progress is recorded, but alignment quietly erodes.
Regulators don’t audit schedules. Their audits demand proof.
When milestones are treated as dates rather than evidence-backed checkpoints, gaps remain hidden until scrutiny begins, during audits, submissions, or due diligence reviews.
When Innovation Outpaces Documentation
One of the most common breakdowns occurs during late-stage design changes. Engineering identifies a necessary improvement and moves quickly to implement it. Documentation, however, doesn’t always keep pace.
Specifications may be updated locally. Risk assessments may not immediately reflect the change. Testing or manufacturing proceeds using earlier revisions that no longer represent the device being built.
This misalignment doesn’t stem from negligence. It’s a structural consequence of disconnected systems that allow development activity and documentation to drift apart.
The impact is significant: invalid test data, repeated studies, delayed readiness, and questions about data integrity that take far longer to resolve than to prevent.

The Audit “War Room” Is a Symptom, Not the Problem
Despite decades of quality maturity, many organizations still experience audits as crisis events. Surprise
inspections or investor diligence requests trigger what quality teams
often call the “war room”—late nights, manual record assembly, and
anxious cross-checking of approvals.
The problem isn’t missing documentation. It’s documentation that’s assembled retroactively.
When records are curated after the fact, inconsistencies surface: approvals dated after dependent activities, risks updated too late, or
test reports referencing outdated requirements. Auditors notice quickly.
An inability to produce a specific record within minutes doesn’t just slow the audit; it invites deeper scrutiny into system reliability.
Source: Enlil
Submission Readiness and the Digital Scavenger Hunt
Regulatory submissions introduce another familiar challenge. As FDA eSTAR becomes standard, regulatory teams are expected to assemble clean, traceable, and current documentation for each submission section.
In practice, this often devolves into a digital scavenger hunt. Multiple versions of “final” documents. Conflicting PDFs. Spreadsheets attempting to reconcile revision histories across tools. Days, or weeks, spent verifying documents that already exist.
The risk isn’t simply inefficiency. Submitting the wrong version can trigger administrative holds or refuse-to-file decisions, creating downstream consequences long after development work is complete.
The Silent Loss of Alignment
For quality and program leaders, the most dangerous breakdowns are frequently invisible. Critical documents sit in draft or pending approval while teams assume progress is being made.
Without shared visibility into readiness, milestones can seem complete when they are not. Alignment drifts quietly, until an external event exposes the issue.
Verbal status updates are no longer sufficient in complex, cross-functional environments. Visibility is the prerequisite for control.
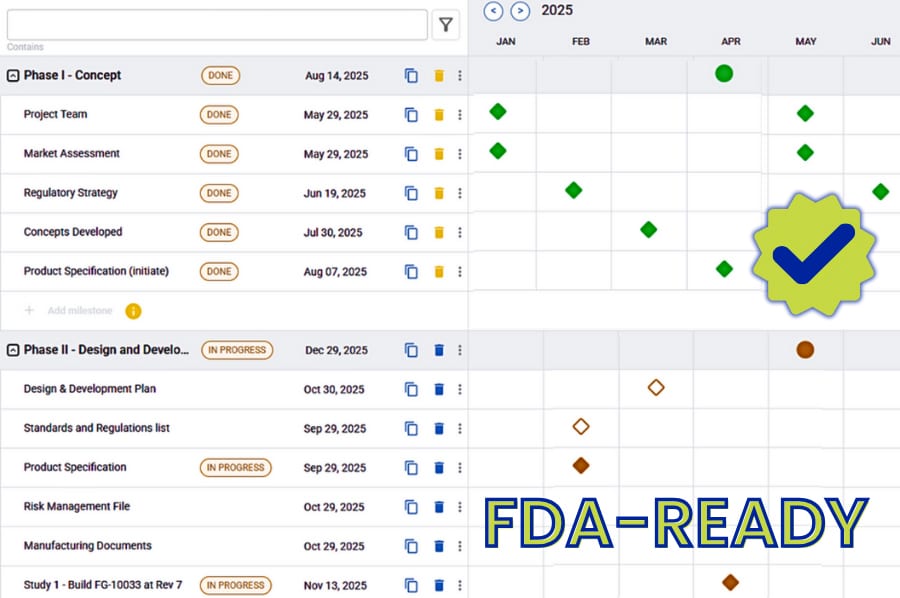
A Proactive Primer for FDA-Ready Milestones
High-performing medical device teams approach milestones differently. Rather than treating them as calendar events, they define them as evidence containers, each milestone explicitly tied to the documentation required to support it.
Proactive teams follow these five recommendations:
- Define milestones by evidence, not activity.
A milestone is only complete when the required documents are approved, current, and traceable. - Lock documentation states at key phases.
At Design Freeze, Verification, and Submission, documentation should be preserved as a snapshot in time, even as future revisions continue. - Align documentation structure to regulatory outcomes.
Organizing documentation the way regulators review it, not the way teams prefer to store it, reduces friction and error during submissions. - Make readiness visible across functions.
Ensure milestone readiness is visible to all functions. Give Engineering, Quality, Regulatory, Operations, and Program Management a shared view to keep alignment on track. - Treat inspection readiness as a daily discipline.
Treat inspection readiness as an ongoing effort. Maintain readiness daily to eliminate last-minute audit preparation.
From Fire Drills to Operational Credibility
The most effective medical device organizations don’t slow innovation to achieve compliance. They design development systems where compliance is a natural outcome of how work progresses.
When milestones are grounded in evidence, audits become routine rather than disruptive. Submissions become predictable. Quality teams move from reactive cleanup to strategic leadership.
In an industry facing increasing complexity, from global supply chains to digital submissions and evolving regulatory frameworks, operational credibility has become the differentiator. It’s the ability to demonstrate, at any moment, not just that work was done, but that it was done correctly, consistently, and in context.
Shifting to milestones that prove readiness ensures medical device teams stay aligned and audit ready.
